





Neben der Technologie war die Synthese von Glykosiden schon immer auch für die Wissenschaft von Interesse, da sie in der Natur eine weit verbreitete Reaktion darstellt. Aktuelle Arbeiten von Schmidt, Toshima und Tatsuta sowie zahlreiche darin zitierte Referenzen erläutern ein breites Spektrum an Synthesemöglichkeiten.
Bei der Synthese von Glykosiden werden mehrere Zuckerkomponenten mit Nukleophilen wie Alkoholen, Kohlenhydraten oder Proteinen kombiniert. Ist eine selektive Reaktion mit einer der Hydroxygruppen eines Kohlenhydrats erforderlich, müssen alle anderen Funktionen im ersten Schritt geschützt werden. Prinzipiell können enzymatische oder mikrobielle Prozesse aufgrund ihrer Selektivität komplexe chemische Schutz- und Entschützungsschritte ersetzen, um Glykoside selektiv in Regionen zu synthetisieren. Aufgrund der langen Geschichte der Alkylglykoside wurde der Einsatz von Enzymen in der Glykosidsynthese jedoch noch nicht umfassend untersucht und angewendet.
Aufgrund der Kapazität geeigneter Enzymsysteme und der hohen Produktionskosten ist die enzymatische Synthese von Alkylpolyglycosiden noch nicht für eine industrielle Umsetzung bereit, und es werden chemische Methoden bevorzugt.
Im Jahr 1870 berichtete MA Colley über die Synthese von „Acetochlorhydrose“ (1, Abbildung 2) durch die Reaktion von Dextrose (Glucose) mit Acetylchlorid, was schließlich zur Geschichte der Glycosidsynthesewege führte.
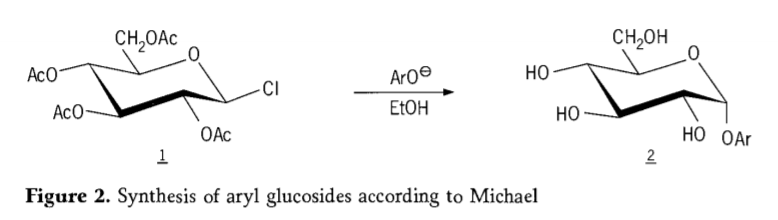
Tetra-O-acetyl-glucopyranosylhalogenide (Acetohalogenglucose) erwiesen sich später als nützliche Zwischenprodukte für die stereoselektive Synthese reiner Alkylglucoside. 1879 gelang es Arthur Michael, aus Colleys Zwischenprodukten und Phenolaten definierte, kristallisierbare Arylglycoside herzustellen (Abbildung 2).
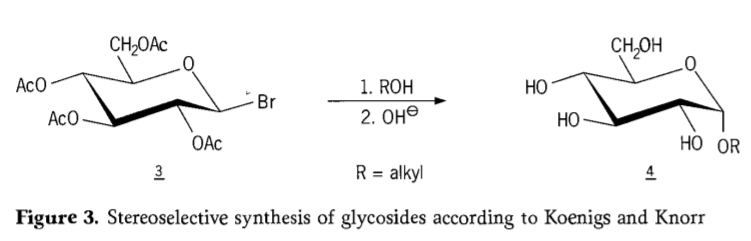
Im Jahr 1901 gelang Michael die Synthese eines breiten Spektrums von Kohlenhydraten und Hydroxyl-Aglyconen, als W. Koenigs und E. Knorr ihr verbessertes stereoselektives Glykosidierungsverfahren vorstellten (Abbildung 3). Die Reaktion beinhaltet eine SN2-Substitution am anomeren Kohlenstoffatom und verläuft stereoselektiv unter Inversion der Konfiguration. So entsteht beispielsweise das α-Glucosid 4 aus dem β-Anomer des Aceobromglucose-Intermediats 3. Die Koenigs-Knorr-Synthese findet in Gegenwart von Silber- oder Quecksilberpromotoren statt.
Im Jahr 1893 schlug Emil Fischer einen grundlegend anderen Ansatz zur Synthese von Alkylglucosiden vor. Dieses Verfahren ist heute als „Fischer-Glycosidierung“ bekannt und umfasst eine säurekatalysierte Reaktion von Glycosen mit Alkoholen. Jeder historische Bericht sollte jedoch auch A. Gautiers ersten Versuch aus dem Jahr 1874 erwähnen, Dextrose mit wasserfreiem Ethanol in Gegenwart von Salzsäure umzusetzen. Aufgrund einer irreführenden Elementaranalyse glaubte Gautier, eine „Diglucose“ erhalten zu haben. Fischer zeigte später, dass Gautiers „Diglucose“ in Wirklichkeit hauptsächlich aus Ethylglucosid bestand (Abbildung 4).
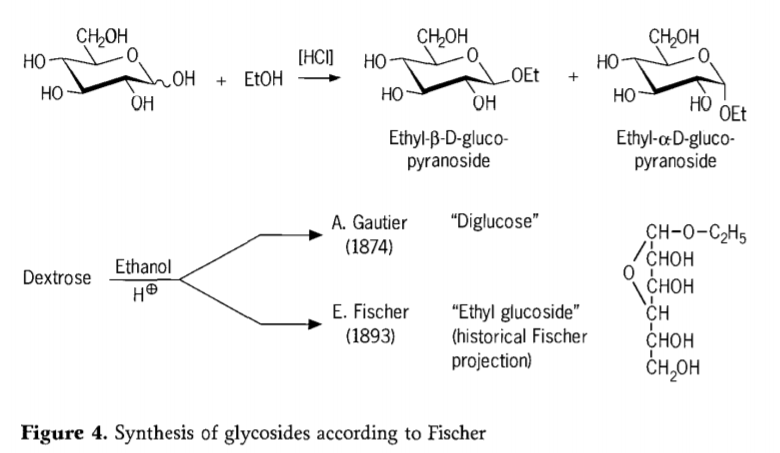
Fischer hat die Struktur von Ethylglucosid korrekt definiert, wie aus der vorgeschlagenen historischen Furanosidformel hervorgeht. Tatsächlich sind Fischer-Glycosidierungsprodukte komplexe, meist Gleichgewichtsmischungen von α/β-Anomeren und Pyranosid/Furanosid-Isomeren, die auch zufällig verknüpfte Glycosidogomere enthalten.
Dementsprechend lassen sich einzelne Molekülspezies nicht leicht aus Fischer-Reaktionsgemischen isolieren, was in der Vergangenheit ein ernstes Problem darstellte. Nach einigen Verbesserungen dieser Synthesemethode übernahm Fischer später die Koenigs-Knorr-Synthese für seine Untersuchungen. Mithilfe dieses Verfahrens berichteten E. Fischer und B. Helferich 1911 erstmals über die Synthese eines langkettigen Alkylglucosids mit tensidischen Eigenschaften.
Bereits 1893 erkannte Fischer wichtige Eigenschaften der Alkylglycoside, wie ihre hohe Oxidations- und Hydrolysestabilität, insbesondere in stark alkalischen Medien. Beide Eigenschaften sind für Alkylpolyglycoside in Tensidanwendungen wertvoll.
Die Forschung zur Glykosidierung ist noch nicht abgeschlossen, und in jüngster Zeit wurden mehrere interessante Synthesewege für Glykoside entwickelt. Einige Verfahren zur Glykosidsynthese sind in Abbildung 5 zusammengefasst.
Im Allgemeinen können chemische Glykosidierungsprozesse in Prozesse unterteilt werden, die zu komplexen Oligomergleichgewichten im säurekatalysierten Glykosylaustausch führen.
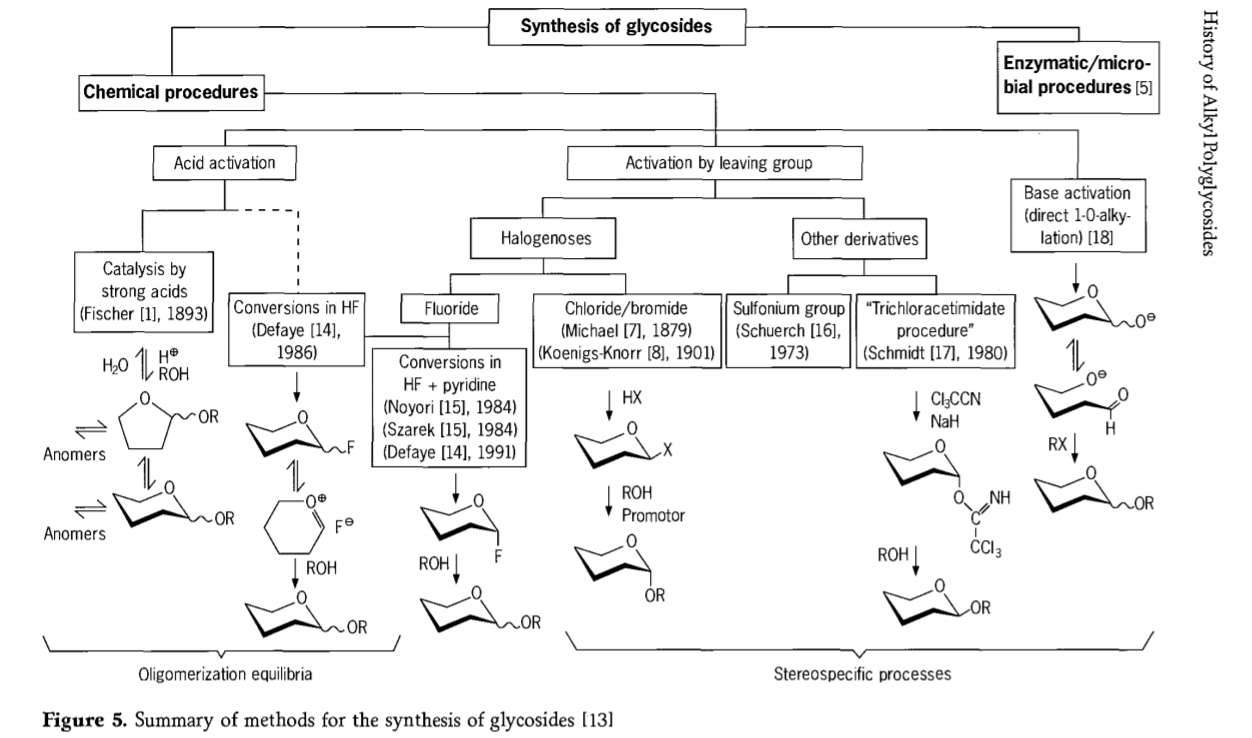
Reaktionen an entsprechend aktivierten Kohlenhydratsubstraten (Fischer-Glykosidreaktionen und Fluorwasserstoff(HF)-Reaktionen mit ungeschützten Kohlenhydratmolekülen) und kinetisch kontrollierte, irreversible und überwiegend stereotaktische Substitutionsreaktionen. Ein zweiter Verfahrenstyp kann zur Bildung einzelner Spezies statt komplexer Reaktionsgemische führen, insbesondere in Kombination mit Konservierungsgruppentechniken. Kohlenhydrate können Gruppen am ektopischen Kohlenstoff hinterlassen, wie Halogenatome, Sulfonylgruppen oder Trichloracetimidatgruppen, oder vor der Umwandlung in Triflatester durch Basen aktiviert werden.
Im speziellen Fall der Glykosidierung in Fluorwasserstoff oder in Mischungen aus Fluorwasserstoff und Pyridin (Pyridiniumpoly[fluorwasserstoff]) werden Glykosylfluoride in situ gebildet und lassen sich, beispielsweise mit Alkoholen, problemlos in Glykoside umwandeln. Fluorwasserstoff erwies sich als stark aktivierendes, nicht zersetzendes Reaktionsmedium; ähnlich wie beim Fischer-Prozess wird eine Gleichgewichts-Autokondensation (Oligomerisierung) beobachtet, obwohl der Reaktionsmechanismus wahrscheinlich unterschiedlich ist.
Chemisch reine Alkylglycoside eignen sich nur für sehr spezielle Anwendungen. So wurden Alkylglycoside beispielsweise in der biochemischen Forschung erfolgreich zur Kristallisation von Membranproteinen eingesetzt, beispielsweise zur dreidimensionalen Kristallisation von Porin und Bacteriorhodopsin in Gegenwart von Octyl-β-D-glucopyranosid (weitere Experimente auf der Grundlage dieser Arbeit führten 1988 zum Nobelpreis für Chemie für Deisenhofer, Huber und Michel).
Im Zuge der Entwicklung von Alkylpolyglycosiden wurden stereoselektive Methoden im Labormaßstab eingesetzt, um eine Vielzahl von Modellsubstanzen zu synthetisieren und ihre physikochemischen Eigenschaften zu untersuchen. Aufgrund ihrer Komplexität, der Instabilität der Zwischenprodukte und der Menge und der kritischen Natur der Prozessabfälle würden Synthesen vom Koenigs-Knorr-Typ und andere Schutzgruppentechniken erhebliche technische und wirtschaftliche Probleme aufwerfen. Fischer-artige Verfahren sind vergleichsweise weniger kompliziert und im kommerziellen Maßstab leichter durchzuführen und werden daher bevorzugt zur Herstellung von Alkylpolyglycosiden im großen Maßstab verwendet.
Veröffentlichungszeit: 12. September 2020